User guide
Steps to follow
1. Input motifs.2. Input query sequences.
3. Input other mandatory inputs.
Reference position
Choose Strand
Threshold frequency
Block size
There are three basic steps to analyze the given query sequences for motifs using MotDet. Following are those steps:
User can enter the custom DNA motifs of length ranging from 5-20. These motifs can be specific pattern or the consensus sequences. Consensus motifs should adher to IUPAC format. Following is the list of symbols used in IUPAC format:
IUPAC Format
|
Symbol
|
Represents what?
|
Why this symbol?
|
|
G
|
G
|
Guanine
|
|
A
|
A
|
Adenine
|
|
T
|
T
|
Thymine
|
|
C
|
C
|
Cytosine
|
|
R
|
G or R
|
puRine
|
|
Y
|
T or C
|
pYrimidine
|
|
M
|
A or C
|
aMino
|
|
K
|
G or T
|
Keto
|
|
S
|
G or C
|
Strong interaction (3H bonds)
|
|
W
|
A or T
|
Weak interaction (2H bonds)
|
|
H
|
A or C or T
|
not-G, H follows G in the alphabet
|
|
B
|
G or T or C
|
not-A, B follows A
|
|
V
|
G or C or A
|
not-T(not U as it stands for uracile), V
follows U
|
|
D
|
G or A or T
|
not-C, D follows C
|
|
N
|
A or G or C or T
|
aNy
|
Input example for custom DNA motif: TATAWAWR,RGWVY
User can also select motifs from the pre defined list. This list is produced after extensive literature search for few mammalian species.
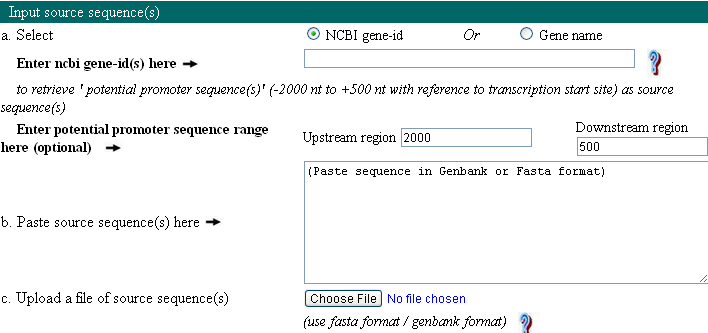
Query sequence should be nucleotide sequences which will be analyzed for user defined motifs. There are four modes to provide this input: Gene ID, Gene name, upload the sequence, and paste the sequence.
Provide NCBI gene ID or gene symbol list separated by comma as an input to analyze their potential promoter sequences. Sequence information is downloaded from NCBI and stored in a local database. User can specify one or more different organism when gene name is provided as an input. This can help user to perform motif analysis of orthogolous genes.
Input example for Gene ID: 23788,7089,283710
Input example for Gene name: MTCH2,TLE2,LOC283710
User can also upload or paste the sequence in FASTA or Genbank format. Click FASTA or Genbank for example. Software works well with query sequence input upto 1 MB.
3. Input other mandatory inputs
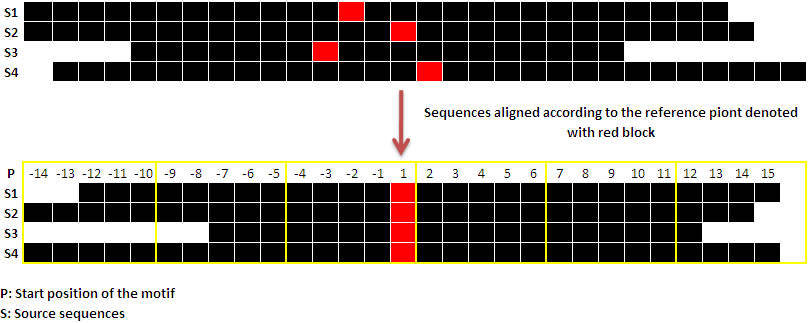
Reference position: As explained in Overview, submitted query sequences are divided into blocks of nucleotides. Before dividing the sequences, they are aligned to each other with respect to some position for e.g. reference point of the gene. Aligning sequences in this manner would help denoting sequencing upstream and downstream uniformly, and hence would aid in block wise analysis of the sequences for motifs. Therefore, reference point plays an important role in blockwise analysis of motif. When user provide gene ID or gene name, transcription start site retrieved from dbTSS will be considered as reference point. And when user upload or paste the sequences, default reference point is the 'Start position' of the sequence which could be altered as per user's requirement.
Following is a pictorial representation, where source sequences are aligned with respect to their reference point. Black lines are the sequences. Reference point is denoted with reb block and yellow lines delimits the blocks of the sequences.
Choose Strand: User can select one strand or both strands for motif analysis. Result for both the positive and negative strand will be given separately.
Threshold frequency: Threshold is defined as minimum no. of source sequences (represented in the form of percentage), in which given motif is expected to be present. Default value of threshold is 40, which can be altered by user if required.
Block Size: To find the common variants and co-variants, sequence will be divided into smaller blocks of sequences. Size of such blocks (in units of base pairs) will be defined using block size. Hence with lower block size, common variants and co-variants will be detected with less mean deviation and vice versa. Default value of block size is 250 nucleotides.
By entering the appropriate inputs, user can carry out blockwise analysis of sequence for the motifs. All the results will be produced on web-browser. To import the result, user can use download link provided at the end of the result page or one has to defined 'Project Name' and 'Email ID' ro recieve result on given email ID.